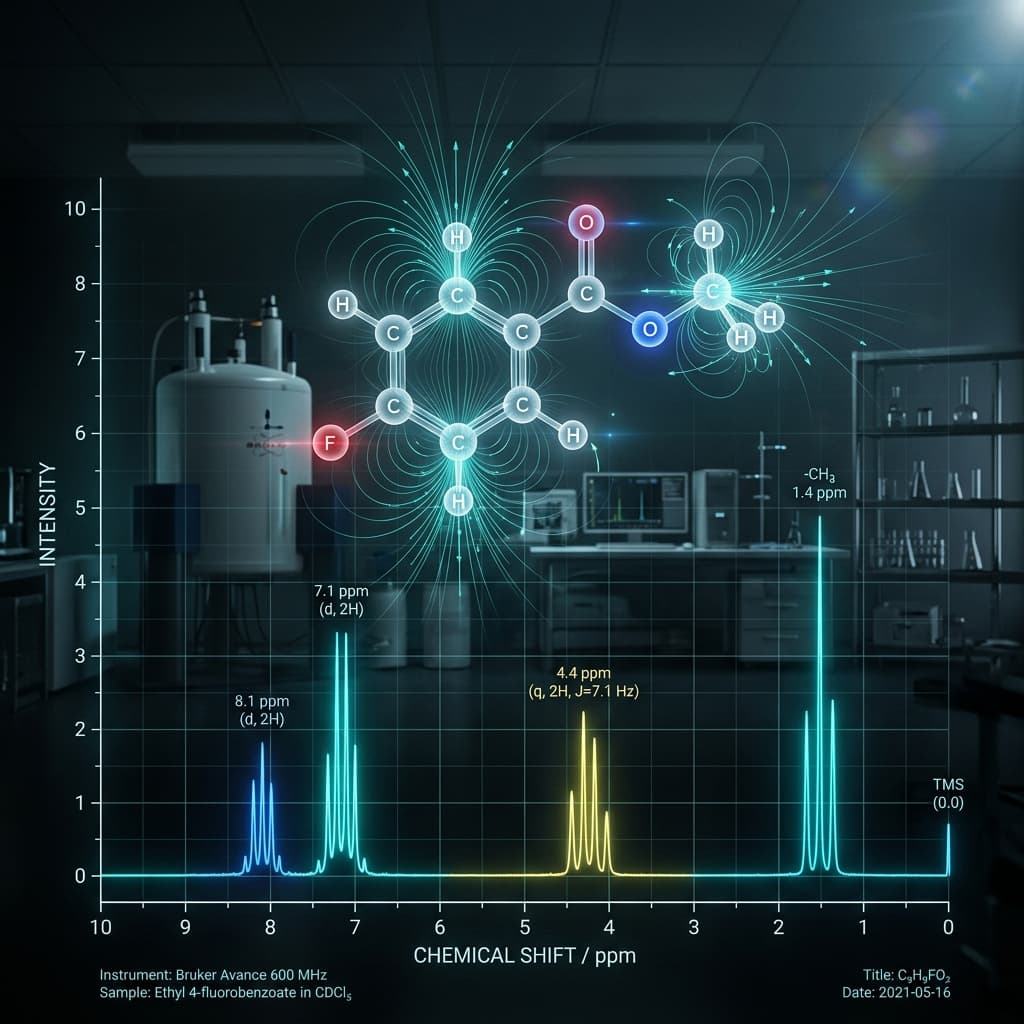
The Language of the Nucleus: Why Shifts Matter
In the world of synthetic chemistry and molecular characterization, ¹H NMR (Proton Nuclear Magnetic Resonance) is the undisputed king. It doesn’t just tell you which atoms are present; it tells you where they are, who they are "talking to" (via coupling), and—most importantly—their electronic environment. That electronic environment is expressed through the chemical shift, measured in parts per million (ppm).
At the Jaconir Team, we encounter thousands of spectra while developing the logic for our NMR Impurity Solver. We’ve seen how a shift of just 0.2 ppm can be the difference between confirming a successful cross-coupling reaction and identifying a failed attempt. Understanding the "why" behind the shift is what separates a technician from a master chemist.
This guide serves as both a definitive 1H NMR Chemical Shift Table and a deep dive into the physical factors that dictate where protons appear on the x-axis of your spectrum.
The Physics of Shielding: Setting the ppm Stage
To understand the table, you must first understand the physics. Every proton in your molecule is surrounded by a cloud of electrons. When you place the molecule in the spectrometer's powerful magnetic field (B₀), these electrons begin to circulate, creating their own tiny, local magnetic field (B_local).
According to Lenz's Law, this local field typically opposes the external field.
- Shielding: The effective field felt by the nucleus is B_eff = B₀ - B_sh. Higher electron density means more shielding, which pushes the signal upfield (to the right, lower ppm).
- Deshielding: Electron-withdrawing groups pull electron density away from the proton. This reduces the opposition to the external field (B_eff increases). The signal moves downfield (to the left, higher ppm).
This simple push-and-pull is the basis for everything in our chemical shift table.
The Definitive ¹H NMR Chemical Shift Table
Below is the standard reference table used by the Jaconir analytical team. These ranges are typical for samples dissolved in CDCl₃ at 298K.
| Functional Group / Proton Type | ppm Range (δ) | Characteristic Appearance | | :--- | :--- | :--- | | Alkanes (Primary, R-CH₃) | 0.8 – 1.0 | Sharp singlet or triplet | | Alkanes (Secondary, R₂-CH₂) | 1.1 – 1.5 | Often complex multiplets | | Alkanes (Tertiary, R₃-CH) | 1.5 – 1.8 | Broad or tiny multiplets | | Allylic (C=C–CHₓ) | 1.6 – 2.6 | Downfield from pure alkyls | | Alkynyl (Terminal C≡C–H) | 2.0 – 3.0 | Sharp singlet (~2.5 ppm) | | Alpha to Carbonyl (C=O–CHₓ) | 2.0 – 2.7 | Distinctive singlets/multiplets | | Benzylic (Ar–CHₓ) | 2.2 – 3.0 | Often overlaps with allylic | | Alkyl Halides (C–I, C–Br, C–Cl) | 2.5 – 4.5 | Moves downfield with electronegativity | | Alkyl Ethers/Alcohols (C–O–H/R) | 3.3 – 4.0 | Strong deshielding from Oxygen | | Vinylic (C=C–H) | 4.5 – 6.5 | Shifted by pi-system anisotropy | | Aromatic (Ar–H) | 6.5 – 8.5 | Complex splitting; Ring Current effect | | Aldehydes (R–CHO) | 9.0 – 10.0 | Highly distinctive singlet | | Carboxylic Acids (R–COOH) | 10.0 – 13.0 | Very broad; highly downfield |
Factors That Drive the Shift: A Masterclass
Why does an aldehyde proton appear at 10.0 ppm while a methyl group sits comfortably at 0.9 ppm? There are four major electronic factors at play.
1. The Inductive Effect (Electronegativity)
This is the most intuitive factor. Atoms like Fluorine, Oxygen, and Nitrogen are "electron-hungry." They pull electron density through the sigma-bonds away from the protons.
- CH₃-Cl: ~3.0 ppm
- CH₃-OH: ~3.4 ppm
- CH₃-F: ~4.3 ppm Notice how the shift increases as the atom becomes more electronegative. This effect falls off rapidly with distance; a proton three bonds away from a Chlorine atom will feel much less "pull" than one directly attached to the Chlorine-bearing carbon.
2. Hybridization of the Carbon
The s-character of a carbon atom affects its electronegativity.
- sp³ Carbon (Alkanes): 25% s-character. Protons are shielded (~1.0 ppm).
- sp² Carbon (Alkenes): 33% s-character. Carbon is more electronegative, deshielding the protons (~5.0 ppm).
- sp Carbon (Alkynes): 50% s-character. Carbon is very electronegative. Wait—why is an alkyne (~2.5 ppm) less shifted than an alkene? The answer lies in the next factor.
3. Magnetic Anisotropy (The Ring Current)
This is the most complex factor. In pi-systems (double bonds, triple bonds, aromatics), electrons circulate in a way that creates a non-uniform (anisotropic) magnetic field.
- Aromatic Rings: The "Ring Current" creates a field that reinforces B₀ on the outside of the ring. This is why benzene protons are pushed all the way to 7.26 ppm.
- Alkynes: The circulation in the triple bond creates a field that actually opposes B₀ along the axis of the bond. This shields the terminal proton, shifting it back upfield to 2.5 ppm.
4. Hydrogen Bonding and Exchange
Protons attached to O or N (like in alcohols, amines, and acids) are highly variable. They participate in hydrogen bonding, which pulls electron density away from the H nucleus.
- In dilute solutions, these peaks shift upfield.
- In concentrated solutions, they shift downfield as hydrogen bonding increases. This variability is why the Jaconir Team always recommends the D₂O shake to confirm these signals.
5. Steric Compression (Van der Waals Deshielding)
In crowded molecules, protons that are physically forced close to other atoms can experience "steric compression." This distorts the electron cloud around the proton, leading to deshielding. If you see a peak that is unexpectedly downfield in a rigid bicyclic system, steric compression is often the hidden culprit.
Advanced Signals: Beyond the Standard Table
Experienced chemists look for signals that don't always appear in a basic ppm table.
Isotopic Satellites (¹³C Satellites)
If you have a very intense, sharp singlet (like the methyls of a t-butyl group), you might see two tiny peaks flanking it. These are ¹³C satellites. Because 1.1% of carbon atoms are Carbon-13 (which has a nuclear spin), the protons attached to them will couple with the carbon nucleus (J_CH ≈ 125–160 Hz). These are not impurities; they are proof of your molecule's isotopic nature.
Paramagnetic Shift Agents
Sometimes, chemists deliberately add "shift reagents" (usually lanthanide complexes like Eu(fod)₃) to a sample. These paramagnetic metals drastically pull signals downfield, often spreading out overlapping peaks so they can be integrated individually. While less common in modern high-field NMR, they remain a powerful tool for complex stereochemical assignments.
Deep Dive: Analyzing Complex Functional Groups
The Aromatic Region (6.5 – 8.5 ppm)
When you look at this region, you aren't just looking for "peaks." You are looking for symmetry and the electronics of the ring.
- Electron-Donating Groups (EDG): Groups like -OMe or -NH₂ push electron density into the ring via resonance. This shields the ortho and para protons, pushing them upfield (closer to 6.5 ppm).
- Electron-Withdrawing Groups (EWG): Groups like -NO₂ or -CN pull density out. This deshields the protons, pushing them downfield (closer to 8.5 ppm).
The Vinylic Region (4.5 – 6.5 ppm)
Protons directly attached to a double bond are shifted downfield from alkanes primarily due to the magnetic anisotropy of the pi-bond. However, the exact position depends heavily on the substitution:
- Terminal Alkenes: Usually appear between 4.8 and 5.2 ppm.
- Internal Alkenes: Often push closer to 5.5–6.0 ppm.
- Conjugated Alkenes: Resonance effects can push these even further downfield.
The Benzylic and Allylic Regions (2.0 – 3.0 ppm)
This area is often "crowded." Protons alpha to a benzene ring or a double bond feel a slight deshielding due to the nearby pi-system. If your molecule has multiple alkyl chains, these benzylic singlets are your "anchor points" for building the structural map.
Case Study: Determining Substitution Patterns
Let’s look at a practical example the Jaconir Team handled recently: distinguishing between ortho-xylene and para-xylene.
- Para-Xylene: High symmetry. The 4 aromatic protons are all equivalent, yielding a single sharp singlet in the aromatic region.
- Ortho-Xylene: Lower symmetry. The protons are not all equivalent, yielding a more complex multiplet (two sets of signals). By combining our ppm table with symmetry analysis, you can determine the exact isomer without needing 2D NMR.
Scan for NMR Impurities in Seconds
Are your shifts not matching the table? You might have residual solvent contamination. Use our interactive solver to compare your experimental peaks against the Fulmer reference database instantly.
Key Experimental Considerations for Precise Shifting
- Referencing Internal Standards: Always check that your TMS is exactly at 0.00 ppm. If not, your entire spectrum is shifted, and the Fulmer tables or our ppm guide won't match. In aqueous samples where TMS is insoluble, use DSS or TSP.
- Solvent Concentration Effects: In some solvents, especially those that can hydrogen bond (like CD₃OD), the chemical shift of certain protons (specifically OH and NH) can drift by up to 0.5 ppm depending on how concentrated your sample is. Always record your concentration for reproducible data.
- Temperature Dependency: Magnetic susceptibility of solvents changes with temperature. If your probe is running hot, your peaks will move. For high-precision work (like variable temperature NMR), internal referencing is non-negotiable.
- Watch the Width: Broad peaks usually indicate exchangeable protons (OH, NH) or restricted rotation (atropisomerism). If you see a broad "hump," try a D₂O exchange or heating the sample to see if the peak sharpens (coalescence).
- Integration is Truth: A shift table gives you the neighborhood, but integration gives you the house number. If a peak is at 4.0 ppm but integrates to 0.1 protons, it's not your ether; it's an impurity.
Conclusion
The ¹H NMR Chemical Shift Table is the primary dictionary of the organic chemist. By understanding the tug-of-war between shielding and deshielding—driven by electronegativity, hybridization, and magnetic anisotropy—you can transform a series of squiggles into a definitive molecular structure.
At the Jaconir Team, we are committed to providing the tools and knowledge that simplify this process. Whether you are using our NMR Impurity Solver to clean up your data or referencing this guide to assign your latest synthesis, remember that every ppm has a story to tell.
Ready to dive deeper? Check out our next guide: How to Tell NMR Impurity Peaks from Real Signals to master the art of spectrum cleanup!
About the Author This guide was produced by the Jaconir Science & Engineering Group. We specialize in building high-precision analytical tools for the modern lab, from mass spec adduct calculators to NMR solvers. Our mission is to bridge the gap between complex spectroscopic theory and daily laboratory practice through elite digital utilities.